
|
Getting your Trinity Audio player ready...
|
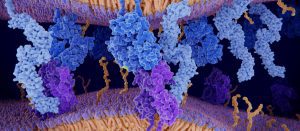 Successful matchmaking with protein molecules is like all other kinds of matchmaking: The two must click for it to work.
Successful matchmaking with protein molecules is like all other kinds of matchmaking: The two must click for it to work.
Except for proteins — the estimated 200 million unique molecular building blocks of life found in all people, animals, plants and bacteria that work together to carry out countless vital functions — figuring out the perfect pair can be a bit complicated.
Compatibility has a lot to do with how they are shaped. It’s like trying to find a specific key to fit a very specific keyhole. Although a difficult and time-consuming process for scientists, knowledge of protein structures and how they best bind is critically important in the design of better medications and vaccines.
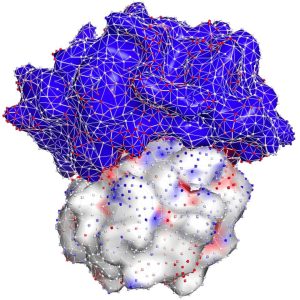
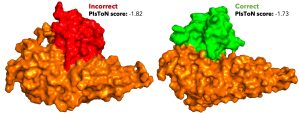
To help narrow the search, a collaborative team of FIU researchers created a new machine-learning model that outperforms similar state-of-the-art software in predicting how protein
molecules will successfully bind together. The AI-based method uses biological and structural information to score the strength of the bond — information that gives scientists a better starting point to figure out how to build the key (in the form of a drug or vaccine) for the lock (the protein). The results were recently published in Nature Machine Intelligence.
“This information is useful in vaccine and drug design,” said the study’s first author Vitalii Stebliankin, who worked on the project as a doctoral student in the Bioinformatics Research Group at FIU. “The first stage of the process is selecting the right ‘candidate’ that would bind to a specific protein molecule out of millions of possibilities. Our framework makes the search faster and more accurate, saving money and resources.”
Why finding the right match is so difficult
The matching process is often so complicated partly because there are so many proteins, all of which are structurally complex.
These little building blocks of life don’t resemble blocks so much as three-dimensional bundles made up of long chains of amino acids that cause them to curl like ribbons or appear as a jumble of tangled wires.

Incredibly flexible, they also wiggle and fold. This movement means they’re capable of coming together in myriad ways — or blocking other molecules’ attempts to connect. In fact, one of the reasons a medication might not be effective is because of a protein’s structure and if it is preventing drug molecules from binding properly.
Putting the protein puzzle together
The three-dimensional structures of proteins long remained a mystery to science — and unraveling it, the holy grail for scientists. After all, shape dictates function. But the process was expensive and required laborious lab work for months — or even years — just to figure out a single protein structure.
Artificial intelligence has led to recent advancements in the field. AlphaFold — a tool created by Google’s subsidiary DeepMind — was one breakthrough, capable of predicting the 3D structure of over 200 million individual proteins.
“AlphaFold was a starting point. We wanted to carry the work forward,” said Giri Narasimhan, a Knight Foundation School of Computing and Information Sciences professor who leads the Bioinformatics Research Group. “We know what the proteins look like in 3D now, but we didn’t have answers on how the proteins interact or where exactly they make contact.”
Narasimhan and his research group teamed with Associate Director of the Biomolecular Sciences Institute Prem Chapagain and molecular biologist Kalai Mathee to provide this missing information and pinpoint the best binding locations.
With a National Science Foundation (NSF) rapid grant, they built the model to incorporate a wealth of research on protein molecules — charge distribution, interactions with water, geometric shape of their surfaces, and where bumps and cavities might be perfect for binding.
It also uses a more effective form of contrastive learning that helps teach the algorithm to make more helpful, nuanced selections.
“Think of contrastive learning this way: If you’re learning to appreciate wine, it’s not enough to know this is a good wine and this is a bad one. Sometimes, it’s also useful to know: Here’s a good wine and then here’s one that’s pretty good but not as good,” Narasimhan said.
“We’re entering an age of rational and effective drug design where we can use a computer to visualize and look at interactions and then design that way: From the ground up.”
— Prem Chapagain
Changing the landscape
A biological physicist, Chapagain has used complex equations to predict how proteins fold. He’s also relied on traditional methods to screen millions of compounds from a database against target proteins, most recently against COVID-19. It can be a fishing expedition.
Computational models and AI could change how he works — for the better, he says. Along with changing the entire landscape of biology and medicine so scientists can make a key to fit the specific shape of a protein molecule.
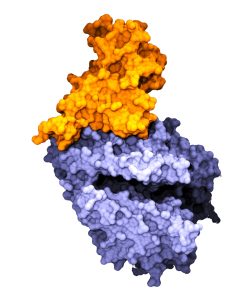
and a human protein hACE2 (bottom).
(Credit: Prem Chapagain)
“We’re entering an age of rational and effective drug design where we can use a computer to visualize and look at interactions, and then design that way: From the ground up,” Chapagain said.
It’s also a step toward opening other doors to personalized treatments and medicines. For example, a headache pain reliever can work effectively on one person and not work at all on another. It’s because we all have minor differences in our protein structures. These genetic differences also exist in different populations that predispose them to diseases or to not be as responsive to certain drugs.
So, what if a drug could be modified to fit the structure? It’s something the team wonders about. It’s not out of the question, though the reality might be a ways off.
For now, they plan to continue their research and continue experimenting with other innovative ways to use AI to benefit scientific breakthroughs and discoveries.




{kind=link}